When is a rash not just a rash? After 30 years of relentless itch, hundreds of stubborn papules, and more than one failed therapy, it’s time to put your diagnostic skills to the test.
Clinical Presentation
A woman in her 40s presented with a 30-year history of an intensely pruritic eruption predominantly affecting the legs, forearms, and hands. She reported a lifelong tendency for skin fragility and sloughing with minimal trauma. She denied mucosal or nail involvement; however, on examination, dystrophy was present in the great toenails and two additional toenails, with the remaining nails appearing normal. There was no clinical evidence of blistering at the time of presentation. Previous therapies, including topical corticosteroids, topical calcineurin inhibitors, and oral antihistamines, had been ineffective. Family history was unknown.
Physical Examination
Examination revealed hundreds of brown to violaceous, 2- to 5-mm, flat-topped, scaly, lichenoid papules distributed on the anterior lower legs, dorsal feet, and dorsal forearms, with some lesions in a linear configuration. Additional scattered lesions were observed on the upper and lower back. Dystrophy of the great toenails and two others was the most notable nail finding. No blisters were present, and the remainder of the examination was unremarkable.
Explore the clinical presentation and diagnostic pathology in the image carousel below:
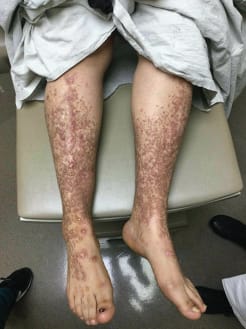
Patient’s lower legs with papules and toenail dystrophy
Hundreds of reddish-brown 2- to 5-mm flat-topped papules on the patient’s bilateral lower extremities and dorsal feet with toenail dystrophy.

Histology, H&E ×10
Basketweave hyperkeratosis, acanthosis, epidermal thinning, and sparse lymphohistiocytic perivascular dermatitis with subepidermal separation and dermal fibrosis.
Diagnostic Workup
Prior biopsies revealed perivascular and focal lichenoid lymphohistiocytic inflammation with milia and a subepidermal separation, suggestive of epidermolysis bullosa acquisita or artifactual separation. A repeat biopsy for hematoxylin-eosin staining showed sparse perivascular to focally lichenoid dermatitis with subepidermal separation and dermal fibrosis, without vacuolar change or eosinophilic infiltration. Additional features included areas of acanthosis, orthohyperkeratosis, and epidermal thinning. Direct immunofluorescence demonstrated no linear deposition of immunoreactants at the basement membrane zone, and a type VII collagen enzyme-linked immunoabsorbent assay was negative. Genetic analysis identified heterozygosity for a c.7247G>T sequence variant in the COL7A1 gene, confirming the diagnosis of dystrophic epidermolysis bullosa pruriginosa (DEB-P).
Clinical Course and Management
Given the findings of lichenified papules on the extremities, severe refractory pruritus, skin fragility, and a subepidermal split on hematoxylin-eosin staining, the diagnosis of DEB-P was supported. Gabapentin provided partial relief, while the addition of dupilumab resulted in significant improvement in pruritus and quality of life.
Discussion
Dystrophic epidermolysis bullosa arises from pathogenic variants in the COL7A1 gene, which encodes type VII collagen—a critical component of anchoring fibrils required for dermoepidermal junction adhesion. DEB-P, first described in 1994, it is a rare subtype that can be inherited in autosomal dominant, autosomal recessive, or sporadic patterns. This variant typically presents with marked pruritus and numerous pruritic papules, papulonodules, and plaques—most commonly on the lower legs and often arranged in a linear configuration. Although scarring, milia, and nail dystrophy may be observed, intact blisters are rarely present. The onset of DEB-P can occur at birth or be delayed by decades, leading to frequent misdiagnosis.
Histopathology frequently demonstrates orthohyperkeratosis, acanthosis, perivascular mononuclear infiltration, and subepidermal separation; milia and dermal fibrosis may also be present. Diagnosis relies on a combination of consistent clinical findings, pathology, family history, genetic testing, and/or immunofluorescence mapping.
Pathogenesis and Treatment
The cause of the severe itch in DEB-P is still not fully understood. Several factors have been proposed, such as abnormal immune signaling involving T-helper cells (including Th1, Th2, and Th17 subtypes), compromised skin barrier function, inflammatory changes linked to ongoing skin injury, and heightened activity of skin nerve fibers that detect itch. Most standard treatments—including antihistamines, medications that calm nerve irritation, topical steroids, and basic wound care—often offer limited relief for the itching and skin discomfort in these patients. For cases that do not respond to typical therapies, drugs like cyclosporine or thalidomide have shown some success. More recently, the biologic medication dupilumab has been reported to significantly reduce itching and improve quality of life in some patients, supporting a possible role for Th2-driven inflammation in this condition.
Differential Diagnosis
In this patient, lichen planus, lichen amyloidosis, and prurigo nodularis were initially considered. However, the absence of dense lichenoid inflammation, vacuolar change, saw-tooth rete ridges, and colloid bodies argued against lichen planus, while the lack of amyloid deposition excluded lichen amyloidosis. Simple prurigo nodularis was considered unlikely due to the convincing subepidermal separation seen on biopsy. Although epidermolysis bullosa acquisita can present with a similar separation and sparse inflammatory infiltrate, the negative direct immunofluorescence, negative serum studies, and lack of blistering did not support this diagnosis. The combination of a pauci-inflammatory infiltrate, subepidermal separation, and genetic findings confirmed DEB-P.
The authors declared having no competing interests.
Source: JAMA Dermatology